Xuran Wang
Welcome to my Homepage!
I’m an Assistant Professor at Seaver Autism Center of Research & Treatment at Mount Sinai.
Before joining Mount Sinai, I worked as a postdoctoral researcher with Kathryn Roeder at Carnegie Mellon University. I earned my PhD degree in University of Pennsylvania in 2019. My thesis “Mendelian randomization and single cell deconvolution: Two problems in statistical genetics” is advised by Nancy Zhang. I also worked with Dylan Small on Mendelian randomization problems and Mingyao Li on statistical modeling of single-cell RNA sequencing.
I received my Bachelor degree of Statistics from University of Science and Technology of China (USTC), School of the Gifted Young.
Here is a current copy of my CV.
Research Interests
My research interests sits in the intersection of statistical genomics, computational biology and neuroscience. I was trained as a statistician and mathematician before diving into the genetics and genomics world. I have broad interests in statistical genetics and genomics, multiomics integration, gene regulatory networks and spatial transcriptomics.
Opening
I am currently looking for postdocs interested in statistical genetics and genomics. Please email a CV to apply.
This is a two year extentable postdoctoral position in Wang’s lab. The successful candidate will apply cutting edge methods in statistics and machine learning to solve scientific problems emerging from genetics, using modern large-scale genomics data such as single-cell sequencing data. We are looking for highly motivated individuals with a strong background in statistical methodology, and a genuine interest in science and data-driven research.
Applicants must have (1) a Ph.D. in statistics, biostatistics, computational biology, computer science or other related quantitative field, (2) strong computing skills, (3) good communication skills.
Contact Me
Constructing local cell-specific networks from single-cell data
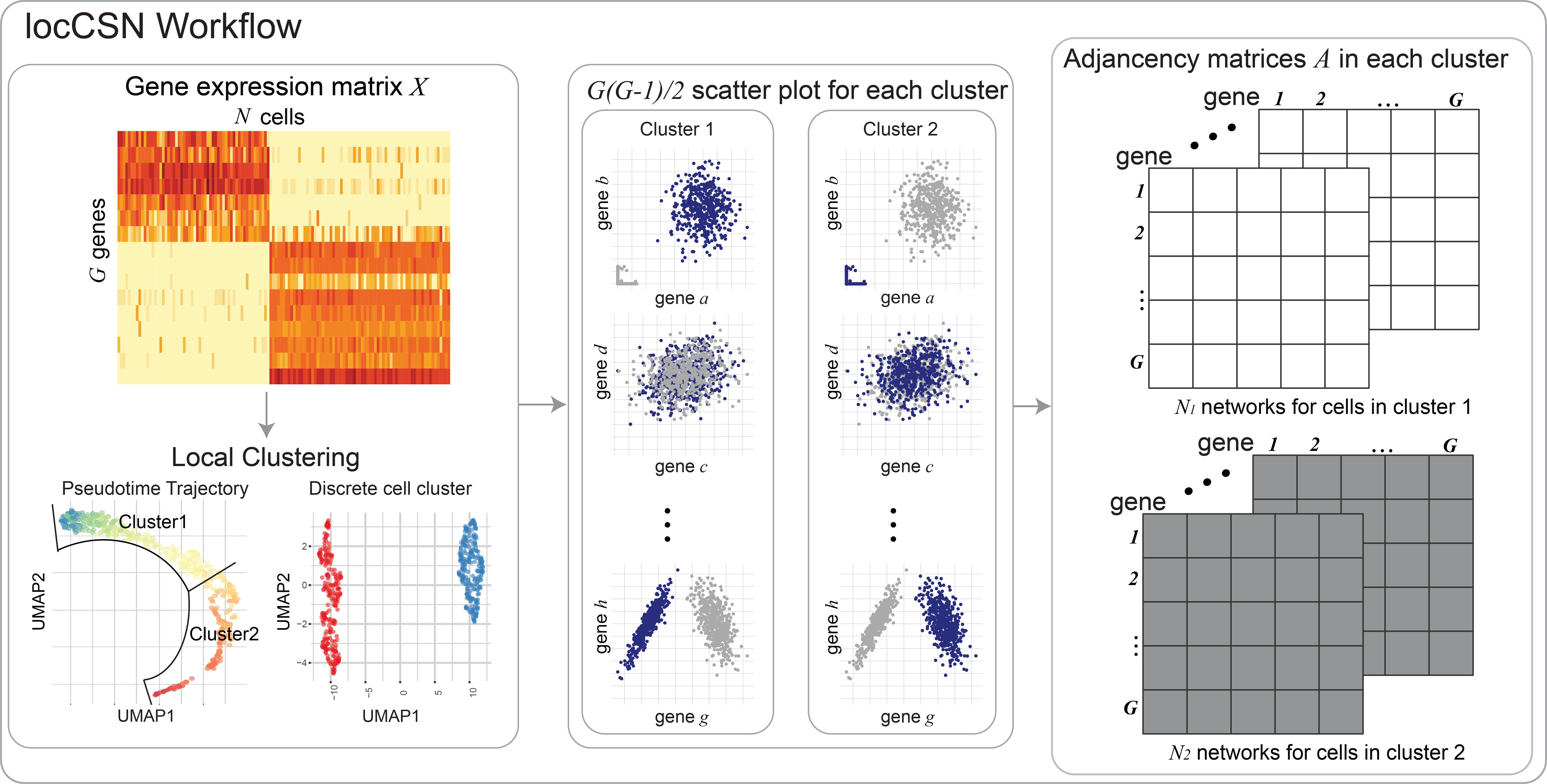
Understanding gene regulatory networks is a topic of great interest because it can provide insights into cellular development, and identify factors that differ between normal and abnormal cells and phenotypes. Single-cell RNA sequencing provides a unique opportunity to gain understanding at the cellular level, but the technical features of the data create severe challenges when constructing gene networks. We develop a method that successfully skirts these challenges to estimate a cell-specific network for each single cell and cell type. Application of our algorithm to two brain cell samples furthers our understanding of autism spectrum disorder by examining the evolution of gene networks in fetal brain cells and comparing the networks of cells sampled from case and control subjects.
Bulk tissue cell type deconvolution with multi-subject single-cell expression reference

Knowledge of cell type composition in disease relevant tissues is an important step towards the identification of cellular targets of disease. We present MuSiC, a method that utilizes cell-type specific gene expression from single-cell RNA sequencing (RNA-seq) data to characterize cell type compositions from bulk RNA-seq data in complex tissues. By appropriate weighting of genes showing cross-subject and cross-cell consistency, MuSiC enables the transfer of cell type-specific gene expression information from one dataset to another. When applied to pancreatic islet and whole kidney expression data in human, mouse, and rats, MuSiC outperformed existing methods, especially for tissues with closely related cell types. MuSiC enables the characterization of cellular heterogeneity of complex tissues for understanding of disease mechanisms. As bulk tissue data are more easily accessible than single-cell RNA-seq, MuSiC allows the utilization of the vast amounts of disease relevant bulk tissue RNA-seq data for elucidating cell type contributions in disease.
Sensitivity Analysis and Power for Instrumental Variable Studies
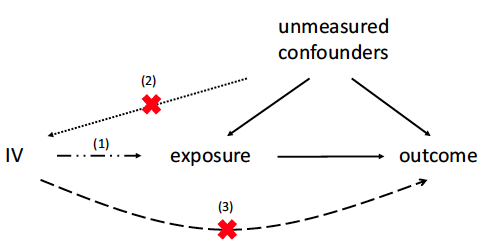
In observational studies to estimate treatment effects, unmeasured confounding is often a concern. The instrumental variable (IV) method can control for unmeasured confounding when there is a valid IV. To be a valid IV, a variable needs to be independent of unmeasured confounders and only affect the outcome through affecting the treatment. When applying the IV method, there is often concern that a putative IV is invalid to some degree. We present an approach to sensitivity analysis for the IV method which examines the sensitivity of inferences to violations of IV validity. Specifically, we consider sensitivity when the magnitude of association between the putative IV and the unmeasured confounders and the direct effect of the IV on the outcome are limited in magnitude by a sensitivity parameter. Our approach is based on extending the Anderson–Rubin test and is valid regardless of the strength of the instrument. A power formula for this sensitivity analysis is presented. We illustrate its usage via examples about Mendelian randomization studies and its implications via a comparison of using rare versus common genetic variants as instruments.
Allele Specific Information in Mendelian Randomization
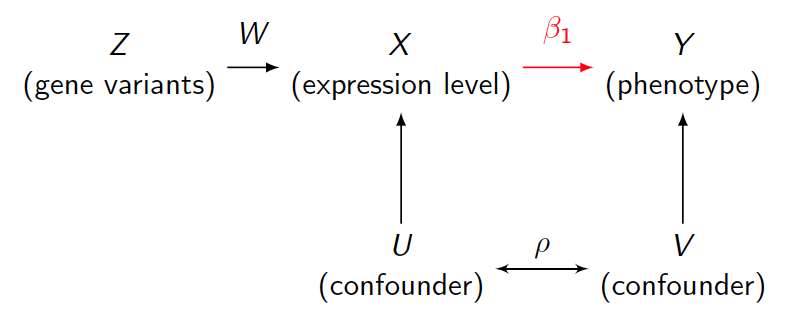
Mendelian randomization (MR) is a method in biomedical research, which uses genotypes as instrument variables to estimate causal effect of exposure to outcome. Conventional estimation is two stage least squares (2SLS). With advanced sequencing technology, allele specific expression information became available. We present a new model to make usage of new information and estimate causal effect using maximum likelihood estimation (MLE). A comparison of precision between 2SLS and MLE is presented with different strength of instrument variables and confounder effect. We also illustrate its usage in finding downstream effect of lncRNA with genotype as instrument.
MuSiC2: cell-type deconvolution for multi-condition bulk RNA-seq data

Constructing local cell specific networks from single cell data

SCEPTRE improves calibration and sensitivity in single-cell CRISPR screen analysis
Detecting cell-type-specific allelic expression imbalance by integrative analysis of bulk and single-cell RNA sequencing data
APOE and TREM2 regulate amyloid-responsive microglia in Alzheimer’s disease
Bulk tissue cell type deconvolution with multi-subject single-cell expression reference

Sensitivity analysis and power for instrumental variable studies

Education & Experience
Icahn School of Medicine at Mount Sinai
Genetics and genomics Science
Carnegie Mellon University
Mentor: Kathryn Roeder
University of Pennsylvania
Thesis Committee: Nancy R. Zhang (Advisor), Mingyao Li, Dylan Small